




Qu’est-ce que la SLA ?
La maladie de Charcot ou sclérose latérale amyotrophique (SLA) est une affection neurodégénérative qui atteint progressivement les neurones moteurs du cerveau et de la moelle épinière. Elle se manifeste dans un premier temps par une faiblesse musculaire, une atrophie des muscles, allant jusqu’à la paralysie progressive. On observe également des difficultés de déglutition, à la parole et des troubles respiratoires. C’est une maladie rare et incurable. Les patients atteints décèdent malheureusement de manière précoce, selon la vitesse de progression de la pathologie.
L’appellation de SLA a été choisie pour décrire les anomalies constatées au niveau microscopique (« sclérose » et « latérale ») ou visibles à l’œil nu (« amyotrophique ») :
- SCLÉROSE : la dégénérescence des motoneurones laisse place à un tissu qui apparaît cicatriciel et fibreux ;
- LATÉRALE : il existe une atteinte des fibres qui proviennent du motoneurone central et qui cheminent dans la partie latérale de la moelle ;
- AMYOTROPHIQUE : la dégénérescence des motoneurones entraîne une fonte des muscles.
Une maladie, plusieurs noms
La SLA, sclérose latérale amyotrophique, est aussi connue sous le nom de « maladie de Charcot ». Le nom de la maladie, Charcot, vient du nom de famille du neurologue français de la Pitié-Salpêtrière, Jean-Martin Charcot, premier à avoir décrit cette pathologie évolutive en 1865, en France. Aux Etats-Unis, on préfère lui donner le nom de Lou Gehrig, un célèbre joueur de baseball, mort de la SLA en 1941.
Qui est touché par la maladie de Charcot ?
Cette maladie des neuromoteurs peut toucher n’importe qui. En règle générale, la SLA est diagnostiquée entre 50 et 70 ans. Une forme très rare de SLA, appelée SLA juvénile, peut se déclarer dans l’enfance. Les premiers symptômes se manifestent alors entre 3 et 20 ans, l’âge moyen du diagnostic étant de 6,5 ans.
- 90 % des formes de SLA sont dites sporadiques, c’est-à-dire que leur origine est inconnue et que tout le monde peut être susceptible de les contracter.
- 10 % des SLA seulement, sont dites familiales ou héréditaires. Dans ces cas-là, la SLA s’est déjà déclarée dans la famille du patient et les mutations génétiques transmises au fil des générations prédisposent les différents membres de la famille à contracter cette neuropathologie.
“A ne pas confondre avec la maladie « CHARCOT MARIE TOOTH »
Pour tout savoir sur la Charcot Marie Tooth
Quelle est l’espérance de vie d’une personne atteinte de SLA ?
L’espérance de vie peut évoluer d’un patient à un autre, mais elle est estimée entre 3 et 5 ans, bien que certaines personnes puissent vivre plus longtemps. Les traitements symptomatiques peuvent faire gagner quelques mois de vie. Il arrive que certains patients vivent jusqu’à 10 ou 15 ans de plus.
Quelles sont les causes de la SLA ?
L’originaire primaire de la SLA sporadique est inconnue
On ne connaît pas la cause initiale des formes sporadiques de SLA. La maladie est probablement multifactorielle, faisant intervenir des facteurs environnementaux et probablement également des facteurs de susceptibilité génétique. Certains facteurs environnementaux ont été suspectés, mais aucun n’a pu être confirmé à ce jour.
Le rôle favorisant d’une activité physique importante ou de traumatismes répétés a été soulevé mais n’est pas prouvé. Quelles que soient les hypothèses, formulées à partir d’études réalisées sur de larges populations de patients, il faut souligner qu’aucune donnée actuelle ne permet de remonter à une cause environnementale dans un cas individuel.
Les causes de la SLA familiale
Les formes génétiques de la maladie représentent un groupe hétérogène. Certaines sont liées au dysfonctionnement d’un gène unique (« formes monogéniques »). C’est dans ces formes que les progrès de la génétique ont permis d’identifier certains gènes responsables. L’anomalie la plus fréquente est une mutation dans le gène de la SOD1 (superoxyde dismutase de type 1) qui n’est toutefois responsable que d’environ 15% de l’ensemble des formes génétiques. D’autres formes correspondent à une hérédité complexe, le développement de la maladie nécessitant probablement la présence d’anomalies de plusieurs gènes (« formes multigéniques »).
Cette forte hétérogénéité explique que devant une forme familiale de SLA, il n’est possible d’identifier une anomalie génétique que dans une petite minorité des cas.
Les mécanismes de la SLA
La sclérose latérale amyotrophique (SLA) est une atteinte des fibres qui proviennent du motoneurone central et qui cheminent dans la partie latérale de la moelle épinière. Localisés dans le système nerveux central et dans la moelle épinière, les motoneurones commandent les muscles volontaires qui nous aident à marcher, à parler et à respirer. Ces muscles responsables de la motricité servent à contrôler les mouvements (par opposition à des muscles comme le cœur, muscle impossible à commander).
Les mécanismes biologiques de la SLA sont de mieux en mieux compris. La maladie résulte d’une cascade d’événements biologiques multiples conduisant à la mort des motoneurones. Beaucoup de progrès ont été réalisés dans la caractérisation des anomalies, notamment grâce à l’analyse de modèles animaux de la maladie.
Les mécanismes de dégénérescence du motoneurone sont interdépendants et ont probablement une
importance relative qui varie en fonction des stades de la maladie. On pense que les anomalies des
mitochondries, les organites cellulaires qui produisent l’énergie nécessaire au métabolisme des motoneurones, jouent un rôle central. L’ensemble de ces mécanismes conditionne le motoneurone à entrer dans une voie finale de mort cellulaire où interviennent des phénomènes d’apoptose (ou « mort cellulaire programmée »).
Infographie – la SLA c’est quoi ?
Les symptômes de la maladie de Charcot
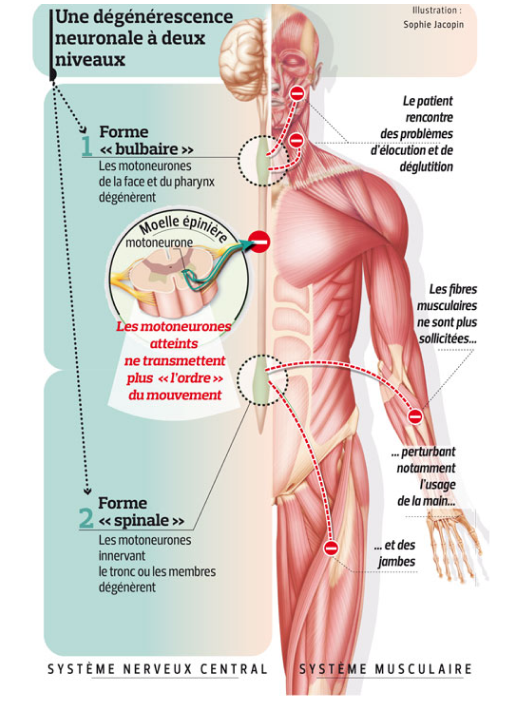
Les signes et symptômes de la maladie de Charcot découlent de l’atteinte sélective des motoneurones commandant les muscles volontaires. Selon le site où débute l’atteinte des motoneurones périphériques, on distingue deux formes de sclérose latérale amyotrophique :
- La forme à début « spinal », liée à l’atteinte initiale des motoneurones de la moelle épinière, entraîne des troubles de la motricité des membres supérieurs ou inférieurs ;
- La forme à début « bulbaire », liée à l’atteinte initiale des motoneurones du tronc cérébral, provoque des troubles de la parole et de la déglutition. Cette forme est plus fréquente chez la femme et débute généralement à un âge plus tardif.
Une caractéristique essentielle de la SLA est qu’en dehors de la motricité, elle respecte les autres fonctions du système nerveux. Les fonctions intellectuelles sont conservées tout le long de la maladie.
La sclérose latérale amyotrophique (SLA) n’affecte également en rien les cinq sens, soit la vision, l’ouïe, le goût, l’odorat et le toucher. Elle n’affecte pas les muscles de l’œil, du cœur, de la vessie, de l’intestin et des organes sexuels. D’autres symptômes peuvent toutefois s’ajouter aux troubles moteurs : constipation, amaigrissement, douleurs, œdèmes et troubles vasomoteurs, troubles du sommeil et troubles respiratoires.
Le diagnostic de la SLA
L’EXAMEN NEUROLOGIQUE ET L’ÉLECTROMYOGRAMME
Le diagnostic de la maladie de Charcot (SLA) peut être difficile, notamment dans les formes de début où les signes sont incomplets. Il n’existe en effet pas de marqueur biologique de la maladie, c’est-à-dire d’anomalie sanguine caractéristique de la maladie. Le diagnostic repose sur l’expertise du neurologue, qui va rechercher des signes d’atteintes du motoneurone central (comme la raideur des muscles, l’exagération de certains réflexes ou la présence d’un signe de Babinski) ou périphérique (comme l’amyotrophie, les crampes, l’abolition de réflexes ou les fasciculations qui sont des contractions involontaires de fibres musculaires visibles à la surface du muscle).
Cet examen clinique est complété par un électromyogramme qui recherche des anomalies électriques de souffrance du motoneurone périphérique et précise leur extension.
Les autres examens sont réalisés au cas par cas, et ont pour but d’éliminer une autre cause pouvant éventuellement expliquer les troubles moteurs. Une IRM du cerveau et/ou de la moelle est souvent réalisée. Une ponction lombaire ou une biopsie musculaire ne sont en revanche nécessaires que dans une minorité de cas.
La prise en charge de la SLA
UNE MALADIE ÉVOLUTIVE
La sclérose latérale amyotrophique (SLA) est une maladie handicapante qui évolue progressivement. Le degré d’évolution de la maladie peut être évalué par des tests cliniques simples et par un bilan respiratoire régulier afin d’évaluer une atteinte éventuelle des muscles respiratoires. Le profil évolutif de la maladie varie en effet d’un patient à l’autre et également chez un même patient au cours du temps. Elle peut se stabiliser spontanément (environ 15 % des patients) à un stade de handicap variable. Il est à priori impossible de prédire l’évolution de cette maladie chez un patient donné.
UN SUIVI MULTIDISCIPLINAIRE
Les patients atteints de SLA doivent être pris en charge par une équipe pluridisciplinaire. La collaboration des différents spécialistes (kinésithérapeute, pneumologue, orthophoniste, psychologue, ergothérapeute,
nutritionniste…) et de différents intervenants (auxiliaire de vie, infirmier…) permet un suivi optimal et coordonné de l’évolution de la maladie.
Les traitements de la SLA
Le traitement de cette pathologie neurodégénérative est essentiellement symptomatique. En effet, étant donné qu’il n’existe pas de traitement curatif à ce jour, l’amélioration du confort du patient dans tous les aspects de sa vie est la priorité. En plus de l’accompagnement des différents spécialistes médicaux, d’autres aides peuvent être apportées :
- Des traitements médicamenteux peuvent être prescrits pour traiter les troubles musculaires (fasciculations, crampes, hypertonie, spasticité), les douleurs, les troubles psychiques, les difficultés respiratoires et de déglutition.
- Des aides techniques peuvent être mises en place afin d’aider le patient à réaliser plus facilement tous les gestes du quotidien, que ce soit à son domicile ou dans son environnement en général.
Depuis octobre 2023, un nouveau médicament, l’AMX0035, a été autorisé en accès compassionnel pré-précoce pour traiter la SLA en France. Ce traitement est toujours en cours d’études aux Etats-Unis et au Canada, mais les premiers résultats indiquent une baisse significative du déclin fonctionnel et une augmentation de l’espérance de vie.